POLYVIEW-3D Tutorial
Part 8. Advanced examples
Below is a set of examples showing how different rendering settings and embedded functions for advanced structure analysis can be used for comprehensive protein structure/function annotation.
Example 1 - Protein interface analysis
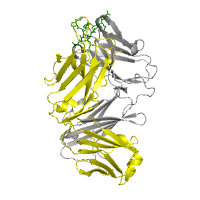
Images below illustrate several specific examples of structure rendering and annotation, using a protein complex of an antibody bound to an antigen derived from Syrian hamster prion protein (PDB id 1cu4). As can be seen from the images, all protein chains can be rendered using different models, including available surface representations (see panels A−D). We furthermore illustrate various structural annotations that can be automatically generated and displayed, including the identification of interacting residues within the complex (panels B and G), distribution of amino acid properties, such as hydrophobicity (panel C) or acidity (panel D), amino acids conservation scores (panels E and F), or putative interacting sites predicted by the SPPIDER server (panel H).
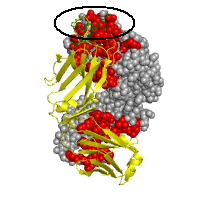
In regard to the latter, we would like to comment that the immunoglobulin complex presented in this example involves two types of interaction interfaces: an obligatory (or permanent) dimerization interface, and a transient epitope-binding interface (circled at panel B). The analysis of the surface of one of the chains reveals a hydrophobic nature of obligatory interface, whereas transient interface has no specific pattern. One can also clearly see that parts of the protein surface that are involved in permanent interactions with the light chain (chain L in 1cu4) tend to be evolutionary conserved, while the interface with an antigen is highly variable, which is consistent with epitope binding.
Consequently, servers that rely on identification of evolutionary conserved and surface exposed residues to suggest functional hot spots, such as ConSurf, are likely to miss the antigen-binding interface. On the other hand, methods that utilize other structural features, including SPPIDER which used here, may be able to identify putative interaction sites even when there is no clear conservation pattern. At the same time, however, induced fit and dependence on structural details may result in lower accuracy in other cases. Therefore, contrasting the results of multiple approaches is likely to increase the confidence in predictions. Using POLYVIEW-3D greatly facilitates such comparative analysis.
| Panel | Image | Annotation | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A |
|
Rendering Chain H (heavy chain) is gray, rendered as cartoon Chain L (light chain) is yellow, rendered as cartoon Chain P (epitope) is green, rendered as wireframe |
||||||||||||||||||||||||||
| B |
|
Interface recognition Chain H: Interface SA=2288Å2, HP index=0.93±0.82, Residues: 31 33 35 37 39 44 45 47 50 52 53 56 58 60 61 91 95 96 98 100 102 119 120 121 122 123 124 125 127 129 134 136 138 140 161 162 163 164 166 168 173 175 177 205 208 210 211 213 Chain L: Interface SA=2210Å2, HP index=0.84±0.87, Residues: 1 27D 32 34 36 38 41 43 44 45 46 49 50 55 87 89 91 92 94 95 95A 97 100 114 116 117 118 119 120 121 122 123 124 127 131 133 135 137 138 158 160 161 162 163 164 165 167 174 176 178 180 207 208 209 211 212 214 Chain P: Interface SA=878Å2, HP index=0.88±0.58, Residues: 105 106 107 108 109 110 111 112 113 |
||||||||||||||||||||||||||
| C |
|
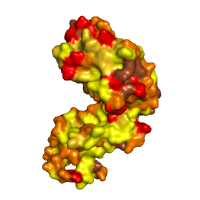
Hydrophobicity profile Hydrophobic (Yellow): A, C, F, G, I, L, M, P, V Amphipathic (Dark yellow): H, W, Y Polar (Orange): N, Q, S, T Charged negatively (Red): D, E Charged positively (Brown): R, K |
||||||||||||||||||||||||||
| D |
|
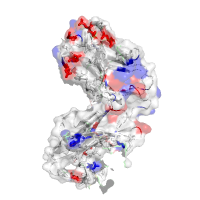
Acidity profile Acidic (Red): D, E Basic (Blue): H, R, K Rest (White): A, N, C, Q, G, I, L, M, F, P, S, T, W, Y, V combined with Interface recognition Chain H: Interface SA=2288Å2, HP index=0.93±0.82, Residues: 31 33 35 37 39 44 45 47 50 52 53 56 58 60 61 91 95 96 98 100 102 119 120 121 122 123 124 125 127 129 134 136 138 140 161 162 163 164 166 168 173 175 177 205 208 210 211 213 |
||||||||||||||||||||||||||
| E |
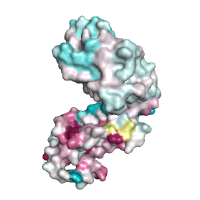
|
Conservation analysis by ConSurf
|
||||||||||||||||||||||||||
| F |
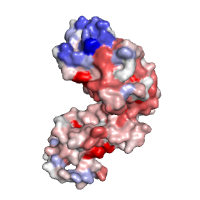
|
Alternative conservation analysis by POLYVIEW
|
||||||||||||||||||||||||||
| G |
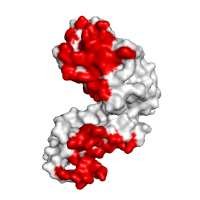
|
Interface recognition Chain H: Interface SA=2288Å2, HP index=0.93±0.82, Residues: 31 33 35 37 39 44 45 47 50 52 53 56 58 60 61 91 95 96 98 100 102 119 120 121 122 123 124 125 127 129 134 136 138 140 161 162 163 164 166 168 173 175 177 205 208 210 211 213 |
||||||||||||||||||||||||||
| H |
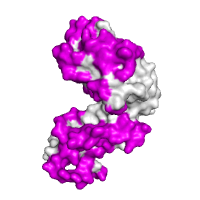
|
Interface prediction Chain H: Residues: 3 21 25 26 27 28 30 31 33 39 41 42 44 45 47 50 52 53 54 58 59 60 61 62 64 65 68 74 91 95 96 97 98 99 100 102 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 136 138 150 153 154 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 177 179 182 208 209 211 212 213 |
||||||||||||||||||||||||||
Example 2 - Protein docking analysis
In this example, we illustrate how POLYVIEW-3D server can be used for protein structure analysis in the context of protein-protein interactions. In particular, we show several specific examples of structure rendering and annotation for a homodimeric complex of regulatory units of the transcriptional antiterminator protein LicT, which regulates the expression of Bacillus subtilis operons involved in beta-glucoside metabolism.
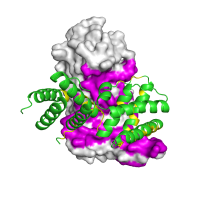
The regulatory units of LicT consist of two five helical bundle domains called PRD1 and PRD2, which adopt dramatically different relative orientations in inactive and active forms of the protein (PDB ids 1tlv, 1h99, respectively). In the activated state, each PRD forms a dimeric unit with its counterpart in the other chain, burying, at the dimer interface, phosphorylation sites that are critical for regulation (conserved histidine residues). In the inactive state, a wide swing movement of PRD2 results in partial opening of the dimer, making the phosphorylation sites accessible on the protein surface. This inactive form of the dimer, with essentially only PRD1 and PRD1' domains involved in the formation of the interaction interface, was used in the CAPRI assessment as target #9, and is shown here in the figure below, panel A.
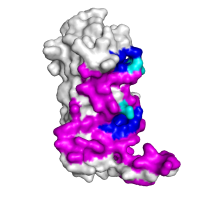
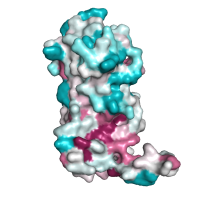
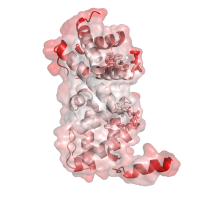
As can be further seen from panels B, C and D, protein chains can be rendered using different models, including available surface representations with different coloring schemes. In particular, we illustrate various structural annotations that can be automatically generated and displayed, including the identification of interacting residues within the complex (panel A and B), visualization of (two) large pockets partially overlapping with the observed interface (shown in blue and cyan in panel B) that were identified using CASTp, coupled with the analysis of evolutionary conservation and putative functional hotspots carried out using ConSurf (panel C), as well as estimated flexibility due to thermal motions, as encoded by temperature factors (panel D).
| Panel | Image | Annotation |
|---|---|---|
| A |
|
Rendering An inactive form of the regulatory domain of LicT antiterminator. The overall structure of the regulatory dimer is shown, with one chain rendered using the surface, and the other chain using the cartoon rendering, respectively. The residues found to be within interaction interface are shown in magenta and yellow. |
| B |
|
Interface and structural pocket identification Chain A of the regulatory domain of LicT protein shown alone, with two largest pockets on the surface that partially overlap with the interaction interface (shown in magenta), as identified using CASTp, highlighted in blue and cyan (the latter for residues within the pockets that are also involved in the formation of the interaction interface). |
| C |
|
Evolutionary conservation Chain A of the regulatory domain LicT protein shown alone, with surface exposed residues colored according to their evolutionary conservation, as assessed by the ConSurf server (residues that are highly conserved are shown in deep purple, whereas highly variable positions are shown in cyan). |
| D |
|
Temperature factors Same chain, with colors representing B-factors this time (red corresponding to highly flexible, and white to relatively rigid parts of the structure, respectively), and with the semitransparent rendering of the surface (residues forming pockets shown in panel B are highlighted using stick models for their side chains). |
| E |
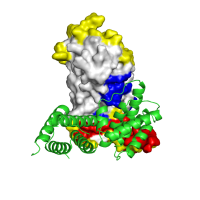
|
Docking model assessment Application of POLYVIEW-3D to the analysis and further assessment (in terms of the overlap between predicted and observed interaction interfaces) of protein docking models, generated for LicT using the ClusPro server. An overall view of the top scoring model is shown, with PRD1 domains forming a qualitatively correct dimer interface. The residues found to be within the interaction interface in this model, which are also predicted by SPPIDER as interaction sites, are highlighted in red, residues observed in the model within interacting sites and not predicted as such are shown in blue, and residues predicted to be interacting sites but not involved in interactions in the model of the complex are shown in yellow, respectively. |
| F |

|
Docking model assessment 2 Visualization of an alternative (and qualitatively incorrect) ClusPro model for the LicT regulatory domain complex, using the same color scheme as in panel E. Note the lack of overlap with SPPIDER predictions in this case. |
In panels E and F, we further illustrate the analysis and scoring of protein docking models. Putative dimer models were generated for this system by the ClusPro server, and were submitted to POLYVIEW-3D. Two different models (ranked by ClusPro as number one and nine, respectively) are shown for comparison. The first model (panel E) is qualitatively consistent with the inactive form of LicT regulatory module, with PRD1 domains forming most of the interface, and PRD2 domains in an open orientation (although somewhat different than observed experimentally - see panel A). The other model chosen here for illustration purposes, and shown in panel F, is characterized by a very different (and incorrect) orientation of the monomers, and the resulting interaction interface.
The correct ranking of these two models by ClusPro is certainly very encouraging. However, in general, multiple and often vastly different models from protein docking simulations are difficult to assess. Therefore, contrasting the results of protein docking approaches with predicted functional hot spots and interaction interfaces, provides a complementary approach to further improve model ranking and confidence in the models selected as top candidates. Using POLYVIEW-3D greatly facilitates such comparative analysis. In this particular case, as can be seen from the figures, only the first model shows significant overlap with predicted (from unbound structures) interaction sites, which are highlighted in red (for residues observed within the interface in a given model, and predicted as interaction sites) and yellow (for the remaining predicted interaction sites).
We would like to comment that SPPIDER predicts in this case two distinct interaction interfaces in the N- and C-terminal regions of the regulatory domain, coinciding with PRD1 and PRD2 subdomains. While only one of these interfaces is present in the inactive form analyzed here in detail, due to the rearrangement of the structure in terms of the relative orientation of PRDs, the other predicted patch overlaps, in fact, with the alternative interface observed in the active form of LicT. The latter can readily be verified using POLYVIEW-3D and the mapping of interaction sites from multiple complexes involving close homologs of the chain of interest.
Example 3 - Enhancing rendered images
Once the image is generated as a static slide, it is possible to
enhance its view as to be used for electronic resources,
such as PowerPoint presentations and Web-pages. On top
of the rendered image at the resulting POLYVIEW-3D page
there is the
Image processing
toolbox that provides an
option for applying different graphic effects to the
original image. Below are examples of available effects
applied to the same protein structure
(PDB id 1n9l) used
in the Ligand and solvent
data section.
| Original image | Transparent (background)* | Reflection* |
|---|---|---|

|
|
|
| Greyscale | X-Ray | |
|
|
|
| Halo(gen) Lighting* | Shadow* | |
|
|
|
| Emboss | Carving | Sketch |

|

|
|
NOTES: (1) Please, be advised that some of the effects (labeled
with asterisk) require
special graphics formats, such as PNG, which makes them
impossible to retain when requested in TIFF or PS
formats for hard-copy publishing. (2) Due to the same
reason, these images cannot be transferred from
web-browser by simple copy/paste or drag-n-drop
operation as being internally processed as bitmaps (at
least, under Windows). Instead, they need to be saved to
local disk first and only then inserted into electronic
document.
(3) The Reflection
effect requires an increase in the image height (1.5
times) in order to have room for 'reflecting surface'.
Last modified: Thu Feb 9 13:31:18 EST 2012